Gary Sharman,a Marcel G. Simonsb and Antony N. Daviesc
aMestrelab Research S.L, Feliciano Barrera, 9B-Bajo 15706, Santiago de Compostela, Spain
bExpert Capability Group - Measurement and Analytical Science (ECG-MAS), Nouryon, Zutphenseweg 10, 7418 AJ Deventer, Netherlands
cSERC, Sustainable Environment Research Centre, Faculty of Computing, Engineering and Science, University of South Wales, UK
I read with interest a recent article in Chemical Science originating out of Jonathan Goodman’s group at the University of Cambridge. Jonathan is another long-standing IUPAC campaigner for scientific data standardisation and his group has been working on an improved solution to tricky nuclear magnetic resonance (NMR) spectra interpretation.1 Their approach exploits modern higher processing speeds to enhance their fully automatic molecular structure elucidation software. Their DP4-AI uses the quantum chemical Gauge-Independent Atomic Orbital Density Functional Theory (GIAO-DFT) method calculations starting from chemical structures with undefined stereochemistry. 1H and 13C-NMR peak picking algorithms handle noisy spectra to predict relative stereochemistry. A statistical value is generated for the likelihood that each of the candidate molecules is correct based on the analysed spectra with almost no need for human intervention. This makes it an ideal tool to rapidly solve difficult problems like natural product library validation.
Clearly, there is still strong demand for improved NMR data interpretation and prediction software. I wondered how much such systems were being used on a day-to-day basis in industry, so talked to Gary Sharman, who has enjoyed a 20-year career in analytical science in the pharmaceutical industry and Marcel Simons, a very experienced NMR expert and one of my old colleagues at AkzoNobel/Nouryon.
Why do automation?
Many years ago, I heard a comment that has stuck in my mind and still raises a smile when I have occasion to remember it. One of the pharmaceutical industry customers of Creon·LabControl AG were testing an innovative combined ultraviolet/visible (UV/vis) and mass spectrometry (MS) automated approach for natural product library screening against “known chemistry” to select extracts for further work. After testing for a while, the customer explained to the software developers the reason behind his excitement. Completely ignoring the technological advances and clever programming that had gone into the system being tested, the customer simply pointed out that the automated spectroscopic data processing system effectively eliminated the boring repetitive work. Extracts that were of no interest (known chemistries) were automatically removed allowing him and his team to very rapidly focus on the extracts of interest that were potential new active molecules. “I can finally spend most of my time doing the expert job my company is actually paying me for”.
So much for the thoughts that people increasing automation might be responsible for taking jobs away from spectroscopists! Gary Sharman highlighted three areas that can be seen as major drivers for better automation:
- Lost opportunities: problems that we would not even dare to start without automation.
- Free up time for more interesting work. We all became spectroscopists for the tricky, interesting problems, not to churn a handle on routine analysis and be bookkeepers. Let automation take care of the drudgery so you can focus on the fun problems. (Like the UV/vis–MS example above.)
- Less silly mistakes/book-keeping errors. We all like to think we are accurate and precise, but the fact is humans make lots of silly mistakes, particularly in collating data. Computers do not make these kinds of mistakes.
Have realistic expectations
The danger of having so much automation at our fingertips is that we might be setting ourselves up for some spectacular falls when the automation encounters problems it simply cannot master. You often see this in much simpler systems such as gas chromatography (GC)/MS database search results of electron ionisation spectra. We have discussed many innovative solutions in this column in the past, but time and again I see reports where the first database hit is cited as being the compound identified—even if the chemistry of the proposed molecule can have nothing to do with what is actually being worked on. If the scientist/student had taken the time to look further down the hit list they would have found a substance that made much more sense in terms of the experiments being undertaken.
So, as Gary put it… If you want perfectly assigned NMR spectra every time—give up now! A much better aim is to really ask yourself what level of errors you are prepared to tolerate, and how that trades off against effort. For example, consider the quality control of a large library; without automation you may conclude it cannot be done. With automation perhaps we have 5 % false positives. It is not perfect, but surely better than having no data on purity.
So, ask yourself what level or errors you are willing to accept. Be realistic. Everyone says “I want 100 % accuracy”, but not even an experienced spectroscopist can achieve that. You might make a trivial error like mixing up two samples or simply working on complex chemistries which you are unfamiliar with.
The automation process
Gary described the process in a similar way to Jonathan Goodman’s group and this actually applies for different types of spectroscopy (Figure 1).
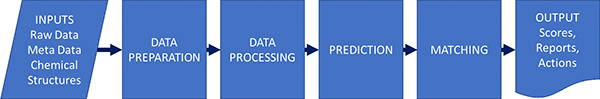
Figure 1. Parts of an automation process; not all processes have all parts. As well as the steps themselves, the inputs and outputs and their interfaces to other systems may be key to success.
Although this might be seen as a rather simple schema, it is good to see how automation will benefit us at the various steps in the process.
- Data preparation and metadata extraction. Not to be overlooked—this may be one of the quick wins. For example, automatically finding and opening connected bits of data, looking up a structure and loading it, saving results—all parts that take time and are tedious bookkeeping, but every process needs them.
- Data processing such as peak picking and categorisation. This can be a very crucial part of the process. Many automated structure validation “mistakes” that are just down to poor peak picking of the data.
- Prediction—unless we are looking up a known thing in a database, we typically must predict the expected result to allow comparison. This could be quite simple (what is the expected ion for MS) or complex (a prediction of NMR by ab initio methods).
- Matching predicted to experimental. For some applications, this may be trivial: is the biggest peak in the mass spectrum the same as the m/z I expect. For proton NMR, with the complexities of coupling, overlap and higher order effects, it is exceedingly difficult.
- Scoring and output—we need to return a useful value that can be used to set actions. We might also want to return “quality factors” that indicate if the result is to be believed or if manual review is a good idea: these two things may well be orthogonal. A fail in the test may not mean the data needs review, and a pass may not mean it is a valid result.
Review by exception strategy
Although you may regard this as an oversimplification, manual analysis is “slow and accurate”. Automation is often seen as “fast but error prone”. By flagging samples for review where there is a reason to believe the automated result may be suspect, we can get the best of both worlds (Figure 2).
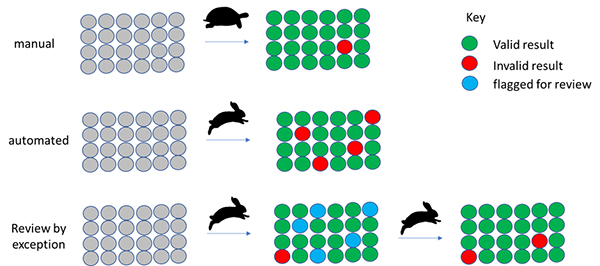
Figure 2. Automation supporting and reducing manual data analysis by focussing on the suspect results.
We do not work alone!
One of the critical questions which we are always asking is exactly how does some new wonder-software fit into our daily working practices and processes?
- The automation steps are only half the problem—how are you going to link your process to other processes in your organisation? This can make or break the automation. Workflow tools like the Swiss KNIME, the Konstanz Information Miner (a free and open-source data analytics, reporting, and integration platform)2 or Biovia’s Pipeline Pilot3 can be valuable here. Also, having information exposed through APIs or web services makes integration easier.
- Constraints. You may have to work with legacy systems, other software with particular requirements or unhelpful interfaces to other data. This can be a major part of the problem that impacts design and implementation.
- The soft part—no one likes to be told by a computer they made a mistake. To get acceptance for a system, it may need thought about how people are informed of failures. For example, an e-mail saying you did something wrong with your boss copied in is probably a bad move. Flagging an error to an expert who reviews it and has a quiet word might be more accepted.
- New problems. Real world data is not perfect. Low signal-to-noise, poorly prepared samples and other components like residual solvents may lead to failures that a person would deal with as part of accepted normal practice.
- Edge cases. Software is built and validated on limited sets of test data. You can guarantee that over time edge cases will be detected that it does not handle well. Hopefully over time, more and more edge cases are dealt with and they become less and less frequent.
So, sticking with the world’s COVID-19 theme, an Automated Structural Verification (ASV) software package like Mestrelab’s “Verify” module can do an excellent job of assigning a molecule, such as a pharmaceutical active ingredient in a clean sample. Expecting a perfect assignment every time may be setting our sights too high. Imperfections do not stop a system being useful.
Enabling non-spectroscopist colleagues
Marcel Simons and colleagues have been working hard to help support colleagues from other disciplines in a speciality chemicals research and manufacturing area in a way that embodies many of the advantages listed above, but in a quite different environment. Their challenges are far more to do with quantitative analysis by NMR rather than purely structure elucidation. The Expert Capability Group’s open shop NMR has to cope with a very high workload of business- and time-critical samples—often being generated out of normal laboratory hours. They started configuring automated spectroscopic data analysis back in 2006. With instrument vendor support, they have developed and deployed over 30 automated methods that do tasks such as data processing for manufacturing plant support. These methods go well beyond the out-of-the box tools, and are designed to work using simple sampling strategies on all liquid samples with usable signals even without the use of deuterated solvents.
The automation results are basically processed spectra and a dedicated Excel file with the desired integrals and calculated molar ratios and/or calculated and normalised weight percentages. Depending on the targeted recipient of the automated processing and the demands of the specific business customers, conditional formatting is applied highlighting the results in green if the processing has delivered the expected result and red if the data is not what was expected and additional actions are potentially required (Figure 3).

Figure 3. At the end of a complex automated NMR data processing method, the customers question may boil down to “is the ratio of the concentration of two compounds within specific target boundaries to the quality criteria”. In this figure, the results show a pass and the lower a fail.
Conclusions
So, it looks like there is a good clear case for continuing to develop faster and less error prone automated spectroscopic data processing. Jonathan’s group have made their new software available under the Open Source MIT license, so if you feel like trying it out while you sit at home worrying about a second COVID-19 wave it can be downloaded from GitHub.4
Gary was one of the authors on a recent paper that pulled together many of the topics discussed here.5 The paper discusses an automated system to verify new compound registrations. At its core was Mestrelab’s Verify engine which automatically verified registered structures against their NMR and liquid chromatography-MS data. This was wrapped in a web service to make access by external processes simple. Bookkeeping tasks, scheduling and interfaces to other systems were taken care of by a KNIME server, and a streamlined review process was put in place to ensure there was a human face put on dealing with any problem samples.
References
- A. Howarth, K. Ermanis and J.M. Goodman, “DP4-AI automated NMR data analysis: straight from spectrometer to structure”, Chem. Sci. 11, 4351–4359 (2020). https://doi.org/10.1039/d0sc00442a
- KNIME. https://www.knime.com
- Pipeline Pilot. https://www.3ds.com/products-services/biovia/products/data-science/pipeline-pilot/
- https://github.com/KristapsE/DP4-AI
- J.A. Lumley, G. Sharman, T. Wilkin, M. Hirst, C. Cobas and M. Goebel, “A KNIME workflow for automated structure verification”, SLAS Discovery (2020). https://doi.org/10.1177/2472555220907091